
")



da Leadership Medica n. 7 del 2003
Un male antico, già presente nel trattato di medicina babilonese compilato tra gli anni 1067-1046 a.C. dove è designato col nome di miqtu (malattia che fa cadere) e dove le sue principali espressioni cliniche sono accuratamente descritte (fig. 1).
Sommario
- Classificazione e forme cliniche
- Eziopatogenesi
- La terapia farmacologica
- Altri trattamenti medici
- Trattamento chirurgico
- Conclusioni
- Bibliografia
N el tempo l’epilessia è stata designata con nomi più o meno immaginifici che riflettono l’atteggiamento della cultura che li ha espressi. Termini descrittivi come quello delle tavolette babilonesi o come falling sickness e mal caduco della cultura medioevale e rinascimentale si alternano con definizioni come male sacro della cultura greca, che alludono ad una supposta origine soprannaturale del disturbo.
el tempo l’epilessia è stata designata con nomi più o meno immaginifici che riflettono l’atteggiamento della cultura che li ha espressi. Termini descrittivi come quello delle tavolette babilonesi o come falling sickness e mal caduco della cultura medioevale e rinascimentale si alternano con definizioni come male sacro della cultura greca, che alludono ad una supposta origine soprannaturale del disturbo.
Di questa visione mitica rimangono tracce nel termine medico epilessia dal verbo greco epilambáno (prendo, invado, afferro) che richiama l’immagine della possessione.
Sarebbe del resto sbagliato pensare che un lungo passato dominato dal pregiudizio sia stato finalmente superato solo grazie ai progressi della conoscenza scientifica moderna: già nel V secolo a.C. Ippocrate contestava appassionatamente il pregiudizio con queste parole “essa” (l’epilessia) “non è a mio parere per nulla più divina o più sacra delle altre malattie, ma ha la stessa natura da cui le altre provengono”.
D’altro canto il progresso scientifico non ha abolito il pregiudizio e lo stigma che circondano questa affezione ancora capace di suscitare in molti una superstiziosa paura, fonte di non necessaria sofferenza per coloro che soffrono di epilessia.
Contro il pregiudizio e la superstizione si battono in tutto il mondo associazioni scientifiche e umanitarie con campagne di informazione, tra le quali merita di essere ricordata la Global Campaign against Epilepsy organizzata e condotta dalla International League Against Epilepsy (federazione delle società scientifiche nazionali di 86 paesi Fig.2) in collaborazione con l’International Bureau for Epilepsy (federazione delle associazioni di volontariato) e con l’Organizzazione Mondiale della Sanità.
Per la medicina moderna l’epilessia è una condizione caratterizzata dalla presenza di episodi accessuali (le crisi) che si ripetono in modo apparentemente spontaneo nel tempo.
Base fisiopatologica ne è la persistente presenza di una eccessiva eccitabilità della cellula nervosa (neurone) che determina la occasionale generazione di scariche epilettiche. Sulla base dei rilievi di prevalenza (6-8% nei paesi sviluppati, fino al doppio nei paesi in via di sviluppo) si calcola che 50 milioni di persone nel mondo soffrano di epilessia.

Classificazione e forme cliniche
Nell’ambito dell’epilessia si distinguono forme cliniche diverse per eziologia, presentazione clinica e prognosi. Se la scarica neuronale epilettogena inizia e si mantiene localizzata ad una popolazione neuronale ristretta, le crisi sono definite parziali ed hanno fenomenologia coerente con le funzioni delle specifiche aree corticali interessate.
Se la scarica inizia localmente, ma si diffonde più o meno rapidamente a vaste aree corticali, vi può essere una generalizzazione secondaria (spesso con manifestazioni convulsive).
Le crisi primitivamente generalizzate sono invece sostenute da una scarica epilettica che fin dall’esordio interessa contemporaneamente ampie aree corticali di ambedue gli emisferi. Molte delle crisi descritte come generalizzate (per lo più di tipo convulsivo) sono in realtà crisi parziali generate da una scarica locale che si diffonde così rapidamente da non permettere l’identificazione di fenomeni parziali iniziali.
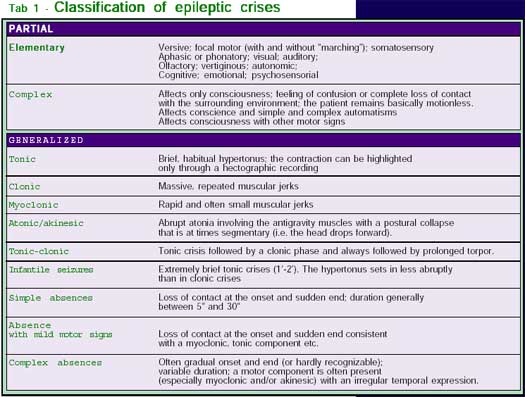
La Tabella 1 riporta la classificazione delle crisi epilettiche elaborate dalla commissione ad hoc della International League Against Epilepsy. Per la classificazione delle crisi e della specifica forma di epilessia hanno valore fondamentale le alterazioni elettroencefalografiche (EEG) concomitanti (EEG critico), molto diverse nelle differenti tipologie di crisi (vedi ad esempio, Fig. 3 e Fig.4).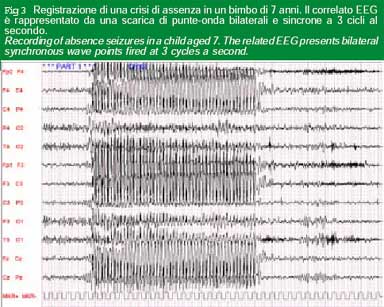
La correlazione tra fenomenologia clinica e correlato EEG può essere analizzata accuratamente in registrazione combinata video-EEG con concomitante somministrazione di semplici test per valutare deficit intra e post-critici.
L’inquadramento diagnostico di una specifica forma di epilessia si basa non solo sul tipo (o sulla associazione di diversi tipi) di crisi presentate dal soggetto ma anche sulle caratteristiche del tracciato EEG intercritico, sull’età di esordio della sintomatologia, sulla presenza-assenza di segni clinici o radiologici di danno del sistema nervoso centrale, sulla presenza-assenza di fattori causali identificabili e sulla famigliarità. Sulla base di questi elementi si giunge a formulare una diagnosi sindromica di forma di epilessia, presupposto fondamentale per programmare ulteriori accertamenti mirati a definirne l’eziologia e a impostare la terapia.
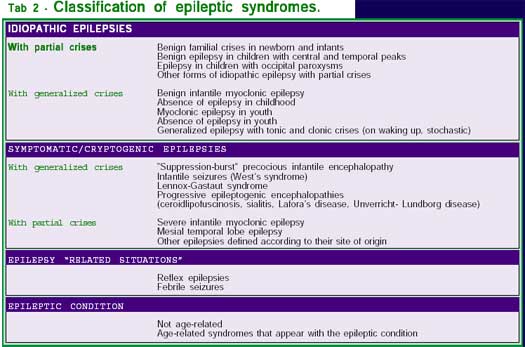
La Tabella 2 riporta una versione semplificata della classificazione delle sindromi epilettiche elaborata dalla International League Against Epilepsy.
Le epilessie idiopatiche sono caratterizzate da esordio legato all’età, normale sviluppo psicomotorio e assenza di danno cerebrale. Lo studio dell’andamento delle anomalie EEG in veglia e in sonno è centrale per la diagnosi e quindi per la prognosi delle differenti forme. Le caratteristiche cliniche delle singole forme sono riportate in dettaglio nei lavori citati in bibliografia.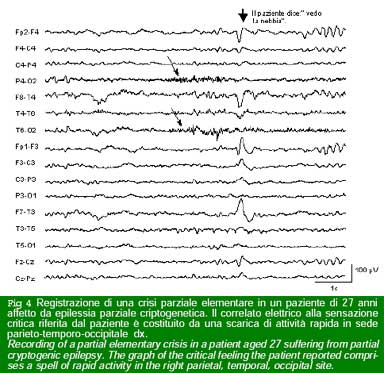 Epilessie sintomatiche/criptogenetiche rappresentano la maggioranza delle epilessie che si protraggono nel tempo e che tendono ad essere resistenti ai trattamenti farmacologici. Possono insorgere a qualsiasi età, comprendono sia forme il cui danno cerebrale può essere individuato e diagnosticato, sia casi in cui esso è solamente ipotizzabile.
Epilessie sintomatiche/criptogenetiche rappresentano la maggioranza delle epilessie che si protraggono nel tempo e che tendono ad essere resistenti ai trattamenti farmacologici. Possono insorgere a qualsiasi età, comprendono sia forme il cui danno cerebrale può essere individuato e diagnosticato, sia casi in cui esso è solamente ipotizzabile.
Le crisi possono essere monomorfe o polimorfe, la fenomenologia ed il quadro EEG dipendono dalla localizzazione del focolaio epilettogeno. Tra le forme parziali un posto particolare occupa l’epilessia del lobo temporale mesiale che presenta un tipico andamento bifasico con un episodio acuto iniziale (spesso una convulsione febbrile protratta) seguito dopo un periodo latente più o meno prolungato, anche di molti anni, da una fase cronica caratterizzata da crisi parziali spesso refrattarie al trattamento farmacologico. Per frequenza e difficoltà di trattamento questa forma rappresenta uno dei maggiori problemi dell’epilettologia dell’età adulta. Le forme generalizzate esordiscono in grande maggioranza in età infantile.
Possono insorgere in assenza di fattori causali noti, in bambini precedentemente normali (forme criptogenetiche), o in presenza di elementi che depongono per un danno cerebrale pregresso, di natura nota o ignota (forme sintomatiche). In questo capitolo si collocano alcune encefalopatie epilettiche dell’infanzia a prognosi severa sia dal punto di vista del controllo delle crisi sia da quello dello sviluppo somato-psichico.
Convulsioni febbrili vengono normalmente definite le crisi che insorgono in fase di salita della temperatura in pazienti al di sotto dei cinque anni di età, neurologicamente sani. Vanno quindi distinte dalle crisi epilettiche vere e proprie che possono essere facilitate dall’ipertermia in pazienti epilettici o portatori di un danno cerebrale.
Eziopatogenesi
Lo studio di modelli sperimentali di epilessia ha evidenziato la potenzialità epilettogena di alterazioni dei meccanismi di eccitabilità neuronale, che dipendono dai flussi di correnti ioniche attraverso i canali della membrana cellulare. Un enorme avanzamento delle conoscenze è derivato dalla recente definizione della struttura molecolare dei canali e dei geni che codificano le proteine che li costituiscono (Fig. 5). Conosciamo oggi con grande precisione le conseguenze di modificazioni strutturali dei canali sulla loro funzione di controllo dei flussi ionici che è regolata da variazioni del potenziale elettrico della membrana (canali voltaggio-dipendenti) o dall’effetto dei neurotrasmettitori (canali associati ai recettori).
Questi stud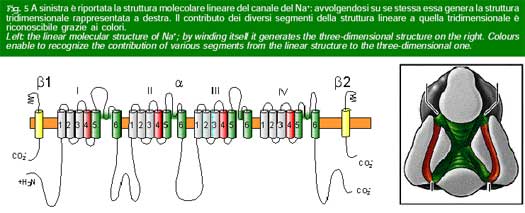 i hanno grandemente avanzato la comprensione dei meccanismi che generano le epilessie e aperto nuove prospettive di trattamento. Le crisi epilettiche possono verificarsi per motivi di pura disfunzione di popolazioni neuronali, in assenza di ogni alterazione morfologica (epilessie idiopatiche). Tuttavia, in un ampio numero di casi, le crisi sono la conseguenza di un danno cerebrale recente o preesistente, accertato (epilessie sintomatiche) o presunto (epilessie criptogenetiche).
i hanno grandemente avanzato la comprensione dei meccanismi che generano le epilessie e aperto nuove prospettive di trattamento. Le crisi epilettiche possono verificarsi per motivi di pura disfunzione di popolazioni neuronali, in assenza di ogni alterazione morfologica (epilessie idiopatiche). Tuttavia, in un ampio numero di casi, le crisi sono la conseguenza di un danno cerebrale recente o preesistente, accertato (epilessie sintomatiche) o presunto (epilessie criptogenetiche).
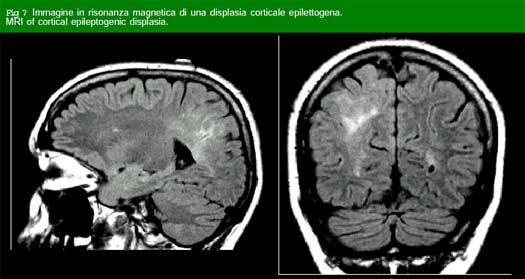
L’evoluzione della diagnostica strumentale neuroradiologica e l’esteso utilizzo della risonanza magnetica, ha permesso di identificare un danno cerebrale misconosciuto in un notevole numero di pazienti. Un esempio importante è l’identificazione di aspetti malformativi localizzati, come le displasie corticali focali (Fig.6).
Per essere efficaci, gli esami di immagine devono essere condotti in modo mirato ad uno specifico quesito posto sulla base della valutazione clinica della forma. Una ulteriore evoluzione è rappresentata dagli esami di immagine funzionale (tomografia a emissione di positroni, risonanza magnetica funzionale, spettroscopia), che hanno importanti applicazioni in casi selezionati (ad esempio, pazienti con epilessia focale candidati per il trattamento chirurgico).
Tra le cause più importanti di epilessia sintomatica sono da ricordare le malformazioni (displasie, lissencefalia, facomatosi, malformazioni vascolari), le encefalopatie fetali e perinatali su base anossica ed emorragica, le encefalopatie infettive e postraumatiche, le vasculopatie (vasculiti, incidenti embolici), i tumori primitivi o secondari del sistema nervoso, le cromosomopatie (trisomia 18, sindrome di Down di Angelmann e di Prader-Willy) e le encefalopatie progressive geneticamente determinate (oltre a quelle riportate nella Tabella 2 le encefalopatie mitocondriali, le acidurie organiche, le aminoacidopati e le malattie perossisomiali. Nelle epilessie idiopatiche non esiste alcun danno cerebrale dimostrabile, una origine genetica è dimostrata o altamente probabile. Le mutazioni responsabili sono state fino ad ora individuate solo in forme rare a trasmissione dominante, che si esprimono con convulsioni ripetute in epoca neonatale o infantile precoce, con convulsioni febbrili o con crisi parziali che persistono in età adulta. 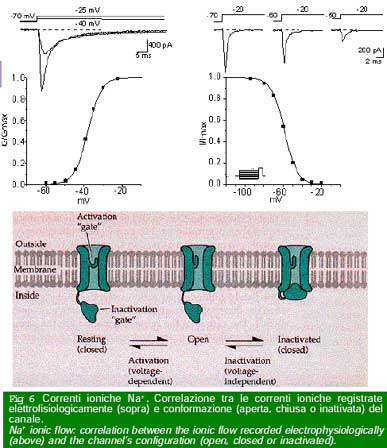
Benché esse riguardino solo una popolazione limitata di pazienti sono di grandissimo interesse in quanto riguardano geni che codificano per subunità di canali ionici voltaggio-dipendenti o associati a recettori. Le mutazioni identificate interessano il recettore muscarinico in forme famigliari di epilessia frontale, i canali K+ nelle crisi benigne neonatali famigliari, i canali Na+ in una forma particolare di epilessia generalizzata con crisi febbrili e il recettore GABA in una sua variante e in una sotto forma di epilessia mioclonica giovanile.
Per le forme più comuni di epilessia idiopatica non si è però giunti alla caratterizzazione molecolare ed è possibile che mutazioni diverse possano determinare fenotipi simili in differenti famiglie o aree geografiche. Per quanto riguarda le epilessie sintomatiche, la disfunzione epilettogena è determinata da alterazioni indotte dalla lesione sulle cellule nervose del tessuto circostante (come nei tumori) o incluse nel tessuto patologico (come nelle displasie). Dati sperimentali ed evidenze indiziarie in epilessie sintomatiche/criptogenetiche umane suggeriscono la possibilità che l’attività epilettica possa di per sé indurre alterazioni del tessuto cerebrale interessato che determinano una progressiva evoluzione peggiorativa dell’area epilettogena fino all’instaurarsi di refrattarietà alla terapia.
Esempio tipico della potenzialità evolutiva del processo epilettogeno è la già citata epilessia del lobo temporale mesiale. L’età del paziente e la sua storia clinica dovranno sempre orientare le indagini diagnostiche. Infatti, la grande maggioranza dei danni cerebrali statici epilettogeni prenatali o perinatali si manifesta spesso con crisi ad esordio precoce, molte encefalopatie “progressive” epilettogene iniziano in una fascia di età relativamente ben delimitata (infantile o giovanile).
Al contrario, alcune lesioni cerebrali acquisite (quali le neoplasie) sono più spesso identificabili nell’adulto. Tuttavia, epilessie ad esordio relativamente tardivo (seconda-terza decade di vita) possono essere causate da mutazioni geniche o da lesioni esistenti fin dalla nascita, quali le malformazioni strutturali focali o le malformazioni vascolari.
La terapia farmacologica
La terapia farmacologica delle epilessie si è arricchita negli ultimi anni per l’introduzione di numerosi farmaci di nuova formulazione progettati per agire specificamente sui meccanismi epilettogeni messi in evidenza dalla ricerca di base.
In particolare tre di essi (oxcarbazepina, gabapentin e lamotrigina) inibiscono la corrente Na +, due potenziano la neurotrasmissione GABA-mediata (vigabatrin e tiagabina) uno, il levetiracetam riduce la corrente Ca2+ e due (topiramato e felbamato) hanno azione multipla su correnti Na+ e neurotrasmissione GABA e Glutammato-mediata. I nuovi farmaci sono utilmente impiegati in tutti i casi di refrattarietà o intolleranza ai farmaci tradizionali. In linea di massima, non vi sono però evidenze che siano più potenti di quelli in uso da decine di anni che sono quindi preferiti come trattamento iniziale perché meglio conosciuti attraverso una lunga esperienza d’uso.
Obiettivo della terapia farmacologica con antiepilettici è quello di sopprimere le crisi o, quando ciò non sia possibile, di limitarne il numero e la gravità, evitando la comparsa di effetti collaterali. Presupposto per il trattamento è la verifica di una tendenza delle crisi a ripetersi nel tempo, che si tratti cioè di una epilessia e non di crisi occasionali.
Una volta posta la diagnosi di epilessia è fondamentale la corretta diagnosi sindromica non solo perché essa determina la scelta del farmaco, ma anche perché la conoscenza della prognosi è importante per la condotta terapeutica. In generale, è opportuno iniziare dopo non più di due tre crisi, una condotta di attesa può essere seguita in forme parziali idiopatiche dell’infanzia (soprattutto nelle forme con crisi rare, in maggioranza notturne) la cui tendenza alla remissione spontanea è certa.
L’inizio tempestivo è particolarmente importante in forme di epilessia criptogenetica e sintomatica, in cui è importante contrastare la tendenza ad una progressiva evoluzione verso la refrattarietà. Non è qui possibile entrare nel merito delle specifiche indicazioni dei singoli farmaci nelle varie forme di epilessia. In generale nelle epilessie idiopatiche il farmaco di prima scelta è il valproato, la carbamazepina può essere usata nelle forme parziali, etosuccimide, lamotrigina e fenobarbitale nei relativamente rari casi di insuccesso del valproato.
Controindicati nelle idiopatiche generalizzate carbamazepina, fenitoina e vigabatrin. Nelle epilessie sintomatiche/criptogenetiche con crisi parziali la prima scelta è per la carbamazepina e tutti gli altri farmaci possono essere usati nei casi relativamente frequenti (50% circa) che non rispondono.
Ancor più difficile è il trattamento delle forme generalizzate sintomatiche/criptogenetiche dell’infanzia che richiedono una specifica competenza. Ricordo solo la specifica indicazione del vigabatrin nella sindrome di West, in cui è tuttavia frequentemente necessario ricorrere agli steroidi. Di regola, il trattamento deve essere iniziato con un solo farmaco e mantenuto con lo schema più semplice possibile. Dopo un periodo di assenza totale di crisi variabile da due a cinque anni si potrà prendere in considerazione la sospensione del trattamento in rapporto a quanto noto sulla storia naturale della specifica forma di epilessia di cui si tratta e ad una attenta valutazione dei fattori di rischio per ricorrenza.
La crisi epilettica non pone, di regola, indicazione a trattamento farmacologico d’urgenza. Il problema si pone solo nel caso in cui le crisi si susseguano a breve distanza o abbiano durata prolungata, configurando uno stato epilettico.
Particolarmente grave è lo stato generalizzato convulsivo che può rapidamente pregiudicare le funzioni vitali del paziente.
I farmaci di primo impiego sono le benzodiazepine, indicate per tutti i tipi di stato di male. Se le benzodiazepine non sono efficaci, può essere utilizzata la fenitoina a dosi di carico e.v. o, se si tratta di una forma generalizzata valproato e.v., se la situazione di emergenza non recede rapidamente con gli antiepilettici è necessario trasferire il paziente in rianimazione e impiegare farmaci o gas anestetici.
Nella definizione dello schema terapeutico sono da tenere particolarmente in conto i parametri farmacocinetici riportati nella tabella e particolarmente quelli rilevanti per la fase di metabolismo-escrezione che è il momento più comune di interazione farmacocinetica fra farmaco antiepilettico e farmaco antiepilettico ed altri farmaci utilizzando se necessario il monitoraggio dei livelli plasmatici da precise indicazioni sul bilancio finale fra assorbimento ed escrezione. Il monitoraggio è necessario in tutte le condizioni fisiologiche (come la gravidanza) o patologiche che possano interferire sull’assorbimento, distribuzione o eliminazione (ad esempio, associazione di farmaci con elevata probabilità di interazione, malattie gravi intercorrenti); il monitoraggio dei livelli plasmatici dovrà essere adattato alle specifiche condizioni cliniche. Poiché solo la quota libera dei farmaci (non legata cioè alle proteine plasmatiche) passa la barriera ematoencefalica, divenendo attiva a carico del sistema nervoso, può essere talvolta necessario monitorare anche la quota libera presso laboratori specializzati. Problemi particolari può porre la condotta terapeutica nella donna. L’efficacia dei contraccettivi può essere ridotta dalla terapia antiepilettica a causa della accelerazione del catabolismo degli estroprogestinici da parte dei farmaci antiepilettici induttori degli enzimi epatici.
In alcune forme di epilessia l’aumento di rischio per crisi in periodo mestruale (epilessie catameniali) può rendere periodicamente insufficiente la protezione farmacologica.
Nelle donne che assumono farmaci antiepilettici la gravidanza deve essere attentamente seguita sia sul piano farmacologico che ginecologico. Il metabolismo di alcuni farmaci (primidone e carbamazepina) può modificarsi in gravidanza con conseguente comparsa di effetti collaterali dovute ad incremento dei livelli plasmatici, che devono quindi essere monitorati con regolarità. Alcuni studi che hanno rilevato un moderato e generico aumento di malformazioni nei nati da pazienti in trattamento con antiepilettici. I maggiori sospetti riguardano il valproato la cui somministrazione correlerebbe invece con una maggior incidenza, rispetto alla popolazione generale, di anomalie a carico delle strutture della linea mediana (variabile dalla spina bifida fino all’anencefalia).
Non vi sono notizie di una specifica teratogenesi per i farmaci di recente introduzione, tuttavia la scarsità delle informazioni disponibili in casistiche umane ne sconsiglia l’utilizzo. In ogni caso il regime terapeutico deve essere razionalizzato prima dell’inizio della gravidanza, verificando che i livelli plasmatici non siano troppo elevati e non abbiano picchi di concentrazione durante il giorno (ciò vale soprattutto per il valproato) e supplementando preventivamente acido folico 4-5 mg/die.
L’uso profilattico di farmaci antiepilettici in presenza di fattori di rischio per crisi (in particolare traumi cranici) non ha dato risultati soddisfacenti.
Discussa è l’opportunità di profilassi farmacologica delle convulsioni febbrili dopo l’occorrenza di un primo episodio vi è invece accordo sulla indicazione a somministrazione estemporanea di benzodiazepine per via rettale in presenza di convulsioni febbrili protratte (durata superiore a 15 minuti).
Per tutti i farmaci antiepilettici sono stati segnalati effetti collaterali a carico del sistema nervoso quali sonnolenza, modificazioni del tono dell’umore, vertigini, atassia, ecc., ed effetti collaterali a carico del sistema gastrointestinale. L’intensità di tali effetti è, in genere, proporzionale alla dose/livello plasmatico dei farmaci antiepilettici (effetti collaterali dose-dipendenti). Un effetto specifico del vigabatrin è la riduzione concentrica del campo visivo, frequente, ma solitamente non avvertita soggettivamente e dovuta ad un effetto del farmaco sulla retina. Inoltre, ogni farmaco può potenzialmente dare, in soggetti predisposti, reazioni idiosincrasiche anche per dosi/livelli plasmatici molto basse (effetti collaterali dose-indipendenti).
Tra le più importanti reazioni idiosincrasiche ad antiepilettici sono da ricordare le rare ma gravi reazioni cutanee (sindrome di Stevens-Johnson e di Lyell) da carbamazepina, difenilidantoina, oxcarbazepina, barbiturici e lamotrigina, l’epatotossicità da valproato e felbamato (anch’essa rara) e le reazioni ematologiche che possono occorrere eccezionalmente con tutti i farmaci, ma che sono particolarmente temibili in pazienti in trattamento con felbamato.
Un soddisfacente controllo delle crisi con minima incidenza di effetti collaterali si ottiene nel 70% dei casi, il restante 30% di pazienti risulta refrattario alla terapia farmacologica. I tentativi di trasferire in campo clinico le esperienze sperimentali indicative della possibilità di prevenire la evoluzione verso una condizione di refrattarietà di alcune epilessie sintomatiche/criptogenetiche con farmaci antiepilettici sono stati fino ad ora deludenti.
Si tratta di un argomento di grande importanza che rappresenta una sfida per la ricerca epilettologica dei prossimi anni. Esistono comunque già ora altri tipi di trattamento medico o chirurgico di cui deve essere considerata l’indicazione nei pazienti refrattari alla terapia con antiepilettici.
Altri trattamenti medici
ACTH e cortisonici: hanno indicazione nella sindrome di West e, più raramente, in forme gravi di epilessia infantile precoce non responsive ai farmaci antiepilettici. Vitamina B6: ha indicazione primaria nel trattamento delle rare epilessie piridossino-dipendenti ad insorgenza infantile precoce.
Dieta chetogena: è basata sulla somministrazione di una dieta ricca di grassi e povera di carboidrati e proteine (con un rapporto 4:1), che induce un aumento dei chetoni ematici. È risultata efficace in soggetti con gravi epilessie farmacoresistenti, in particolare in età infantile. Immunoglobuline: dati non omogenei di efficacia riguardano l’uso di immunoglobuline. In forme gravi, farmacoresistenti e con elevata frequenza critica.
Plasmaferesi: è stata utilizzata in pazienti affetti da epilessia parziale continua associata a encefalite di Rasmussen tipica o atipica. Mira a rimuovere frazioni anticorpali a possibile effetto citotossico o eccitotossico (anticorpi antirecettore per il glutammato).
Trattamento chirurgico
Nelle epilessie sintomatiche di lesione cerebrale che ponga di per sé l’indicazione all’intervento (lesioni evolutive o malformazioni vascolari a rischio di sanguinamento) questo è generalmente mirato alla asportazione della lesione e non all’eventuale attenuazione della sintomatologia critica. Nelle epilessie con crisi focali associate a danno cerebrale non evolutivo o in cui nessun danno cerebrale sia rilevabile, l’indicazione a intervento, mirato al controllo delle crisi attraverso la eliminazione chirurgica dell’area epilettogena, può essere considerata quando sussistano le seguenti premesse.
1) Le crisi si sono dimostrate resistenti ai farmaci indicati per le epilessie parziali;
2) vi è la certezza (o una elevata probabilità) che tutte le crisi inizino a carico di una stessa area cerebrale (area epilettogena);
3) si può prevedere che l’asportazione dell’area epilettogena non causi deficit neurologici significativi.
Tali premesse devono essere rigorosamente verificate attraverso uno studio mirato di ordine clinico-neurofisiologico, neuropsicologico e neuroradiologico.
Lo studio clinico-neurofisiologico deve comprendere la registrazione di crisi mediante monitoraggio prolungato Video-EEG per l’esatta definizione topografica.
Quando l’insieme dei dati clinici, EEG, neuroradiologici e psicologici non risolvano il quesito della precisa sede di origine o pongano il sospetto di una multifocalità, è necessaria una esplorazione con elettrodi di profondità da effettuarsi in ambiente altamente qualificato. Con queste premesse i risultati della terapia chirurgica dell’epilessia sono ottimi e possono consentire il controllo completo della sintomatologia critica nella maggioranza dei pazienti con epilessie parziali refrattarie alla terapia farmacologica.
L’intervento di emisferotomia può essere preso in considerazione in alcuni casi di epilessia infantile associata a deficit neurologici gravi (emiplegia o emiparesi grave) controlaterali all’emisfero leso. Con maggior cautela va considerata la tecnica “palliativa” basata sulla callosotomia parziale, mirata non ad eliminare la scarica epilettica ma a limitarla, in soggetti con epilessie molto gravi. In un limitato numero di pazienti refrattari non candidabili alla terapia chirurgica di elezione può essere considerata l’indicazione a interventi palliativi: callosotomia, tecniche di stimolazione intracerebrale o del nervo vago.
Conclusioni
In pochi campi della medicina l’integrazione tra scienza di base ed esperienza clinica è stata così stretta ed efficace come nell’epilettologia.
Grazie alla accurata osservazione della fenomenologia clinica e agli avanzamenti della neurofisiologia, della biologia molecolare e della farmacologia il neurologo dispone oggi di potenti armi farmacologiche e chirurgiche con cui risolve efficacemente il problema nella larga maggioranza dei pazienti.
Rimane aperto il problema dello sviluppo di nuovi antiepilettici per il controllo delle crisi refrattarie alle terapie e in termini generali quello della identificazione di nuove strategie “antiepilettogene” atte a prevenire l’instaurarsi del processo epilettogeno o a modificarne il corso.
E’ quanto ci aspettiamo dalla ricerca dei prossimi anni sottolineando che non si potrà essere soddisfatti dei progressi della medicina se essi non si accompagneranno ad una modificazione del pregiudizio e delle dinamiche di esclusione che ancora gravano sulle persone che soffrono di epilessia.
Giuliano Avanzini
Direttore Dipartimento di Neuroscieze cliniche Istituto Nazionale Neurologico C.Besta. Milano
Presidente della International League Against Epilepsy (ILAE)
Bibliografia
1. Wilson J.V.K., Reynolds E.H. Translation and analysis of a cuneiform text forming part of a Babilonese treatise on epilepsy. Medical History 1990; 34: pp.185-198. Ippocrate. La malattia sacra. Boringhieri, Torino 1961; pp. 37.
2. Satishchandra P., Gururaj G., Mohammed Q.D., Senanayake N., Silpakit O. From prejudice to hope. Global Campaign against Epilepsy: Out of the shadows. World Health Organization. 2001;pp.62.
3. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989; 30: 389-399.
4. Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 1981; 22: 489-501.
5. Engel J.Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001; 42: 796-803.
6. Avanzini G., Franceschetti S. Cellular biology of epileptogenesis. The Lancet Neurology 2003; 2(1) 33-42.
7. Mody I. Ion channels in epilepsy. International Review of Neurobiology 1998; 42: 199-225.
8. Berkovic S., Scheffer IE. Genetics of the epilepsies. Curr Opin Neurol 1999; 12:177-182.
9. Ben-Ari Y., Represa A. Brief seizure episodes induce long-term potentiation and mossy fibers sprouting in the hippocampus. TINS 1990; 13: 312-318.
10. Spreafico S, Avanzini G, Andermann F. (eds). Abnormal cortical development and epilepsy. Mariani Foundation Paediatric Neurology Series: 7, John Libbey 1999; pp324.
11. White HS. Clinical significance of animal seizure models and mechanism of action studies of potential antiepileptic drugs. Epilepsia (Suppl 1) 1997; 38: S9-S17.
12. Perucca E. Clinical pharmacology and therapeutic use of the new antiepileptic drugs. Fundam Clin Pharmacol 2001; 15: 405-417.
13. Battino D., Dukes G., Perucca E. Anticonvulsants. In: Meyler’s side effects of drugs. (14th Edition).
14. Dukes MNG, Aronson JK (Eds),. Elsevier Science B.V., Amsterdam 2000; 164-197.
15. Löscher W. Animal models of intractable epilepsy. Prog Neurobiol 1997; 53:239-258.
16. Walker MC., White HS, Sander JWAS. Disease modification in partial epilepsy. Brain 2002; 125: 1937-1950.